

Create and organize an SRA submission package¶
Important
A Reminder on File Names and other SRA Requirements
NCBI has extensive requirements for depositing data into the SRA. The CyVerse submission pipeline is an attempt to streamline this process, but your submission must meet several requirements. File names must be unique, and may not contain special characters (e.g. { } ? * . , etc.) or spaces.
The SRA Submission Quick Start is the authoritative guide to SRA requirements. It is worth reading through this before submission.
Important
This quickstart assumes you have uploaded your files to the CyVerse Data Store. If not, following the directions for uploading files to the Data Store. If possible, you may wish to compress these files using gunzip/bzip2 before upload.
(Optional) Compress files in the CyVerse Data Store using gunzip¶
For submission through CyVerse, the sequence files must be compressed. If your files are compressed (.gz/.bz2) you may skip this step.
- Login to the CyVerse Discovery Environment
- Click this link to open the Compress files with gzip App or click the Apps button and search for the “Compress files with gzip” App.
- Under “Inputs” select the individual file (FastQ/SFF/BAM) to compress
- Click ‘Launch Analysis’ to compress the file and click the Analysis button to monitor job status and view results. Once all files are compressed you may wish to gather them into a single folder to begin your submission.
Tip
Each file (e.g. read_R1.fastq) must be individually compressed (e.g. read_R1.fastq.gz)
Create SRA submission package and add sequence data¶
An SRA submission requires that your sequencing data are organized in a specific structure of folders and subfolders. It will be helpful if you are familiar with SRA terminology:
Tip
SRA Terminology
Adapted from NCBI:
- BioProject: A BioProject is a collection of biological data related to a single initiative, originating from a single organization or from a consortium.
- BioSample: The BioSample database contains descriptions of biological source materials used in experimental assays. Different experimental conditions, tissue types, etc. would typically be different BioSamples.
Your submission will contain the following folders and data:
Folder Description Contents Metadata BioProject (One per submission) This is the top-level folder and will contain all other folder and data. One or more BioSample folders A BioSample Metadata template will specify details like the project summary, submitter/investigator information, etc. BioSample (One or more per submission) BioSample folders, one for each experiment/ tissue type, etc. One or more library folders A species/tissue specific metadata template will describe biological information about the sample (e.g. sex, collection location, etc.). Library (One or more per submission) These folders will contain the sequencing data. Each replicate will have its own library folder. Each library folder will contain one either one (single-end) or two (paired-end) sequence files. A library metadata template will describe information about the sequencing run
Login to the CyVerse Discovery Environment.
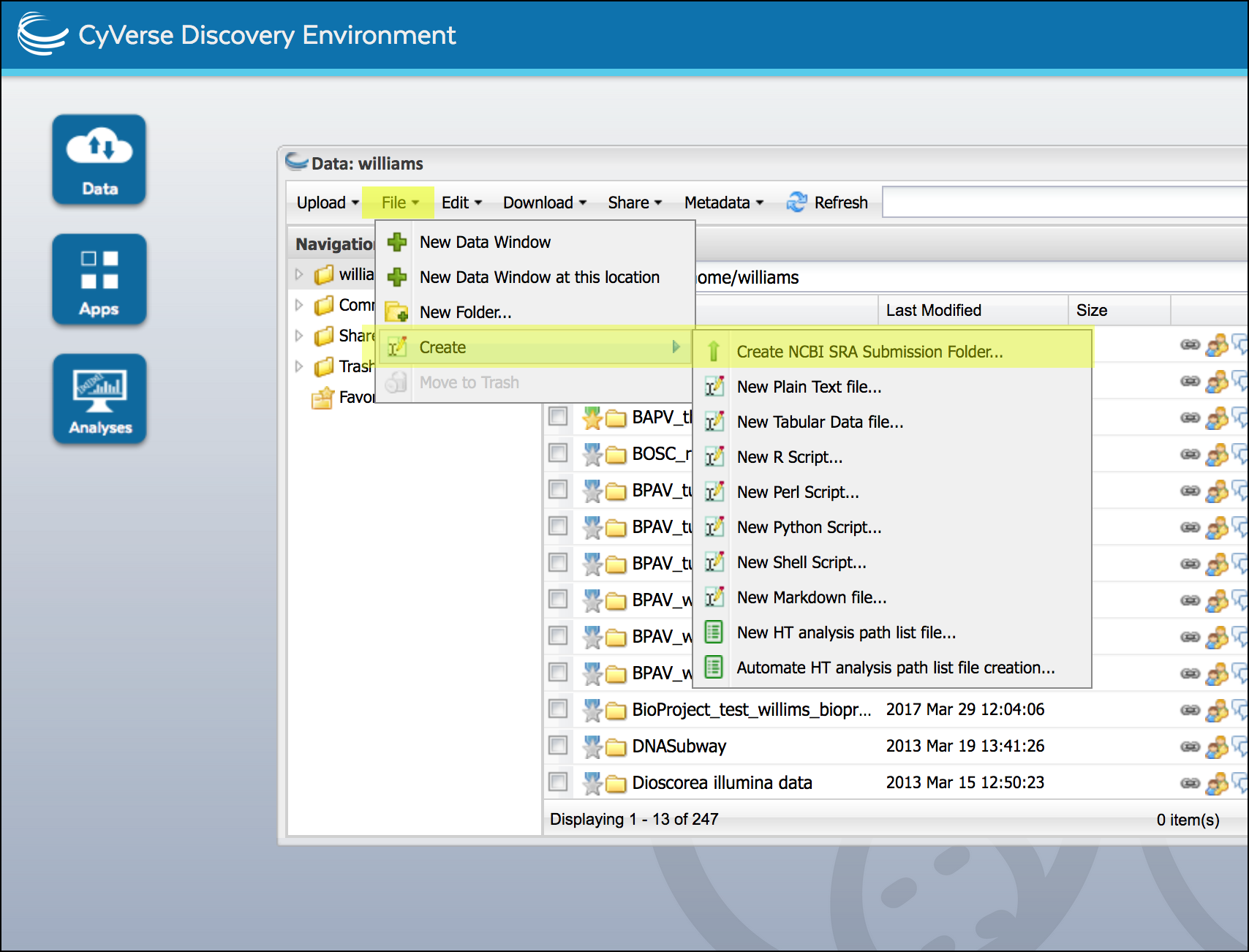
Click on Data to open a data window. In your home directory (or any directory your own) create the submission folder; Click on the “File” menu, then click ‘Create’ and select ‘Create NCBI SRA Submission Folder’.
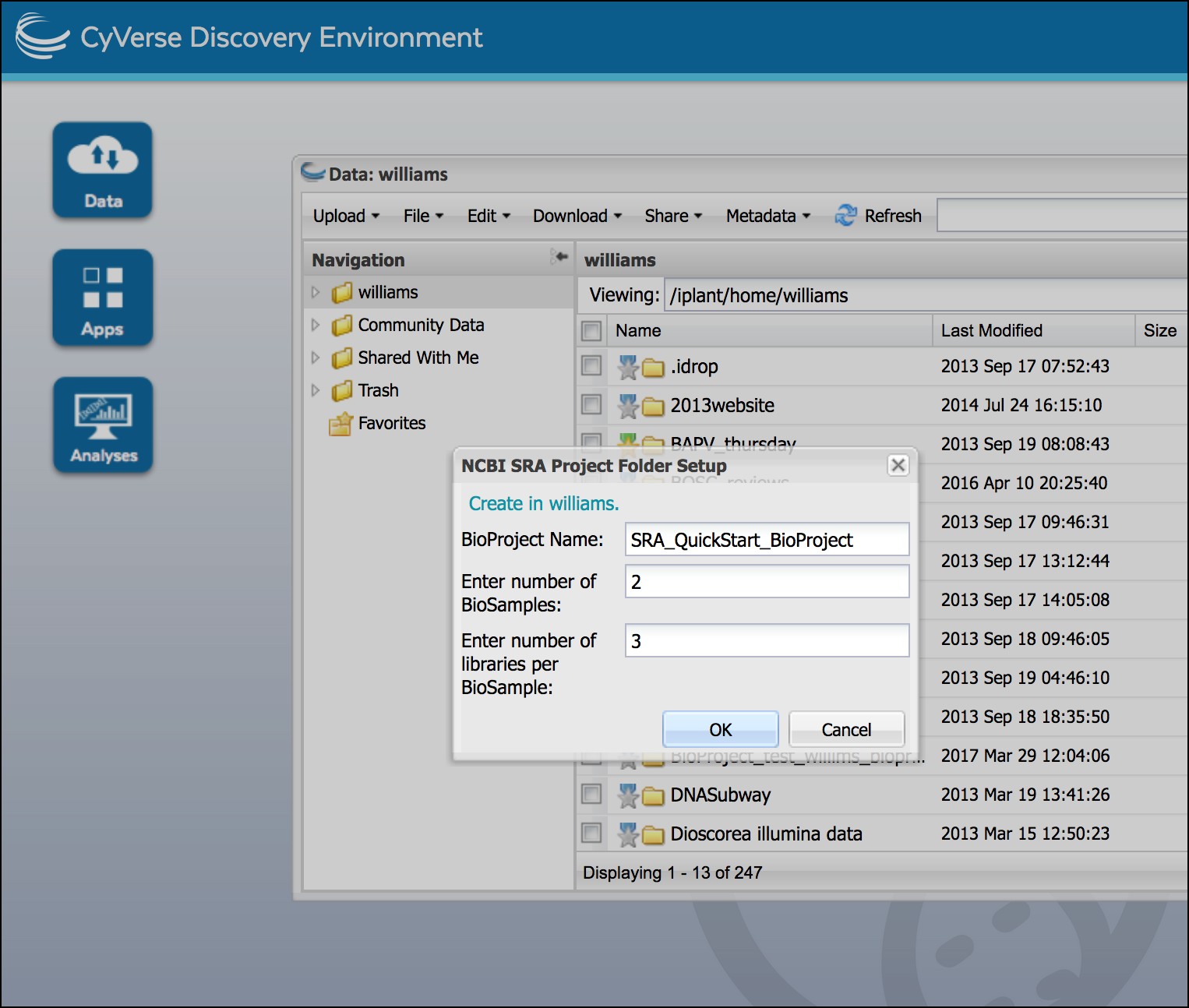
Enter the name of your BioProject, and the number of BioSamples and libraries. click ‘OK’ to create the submission folders.
Tip
In our Example data We have two experimental conditions with 3 sequencing replicates each so 2 BioSamples and 3 libraries in each BioSample
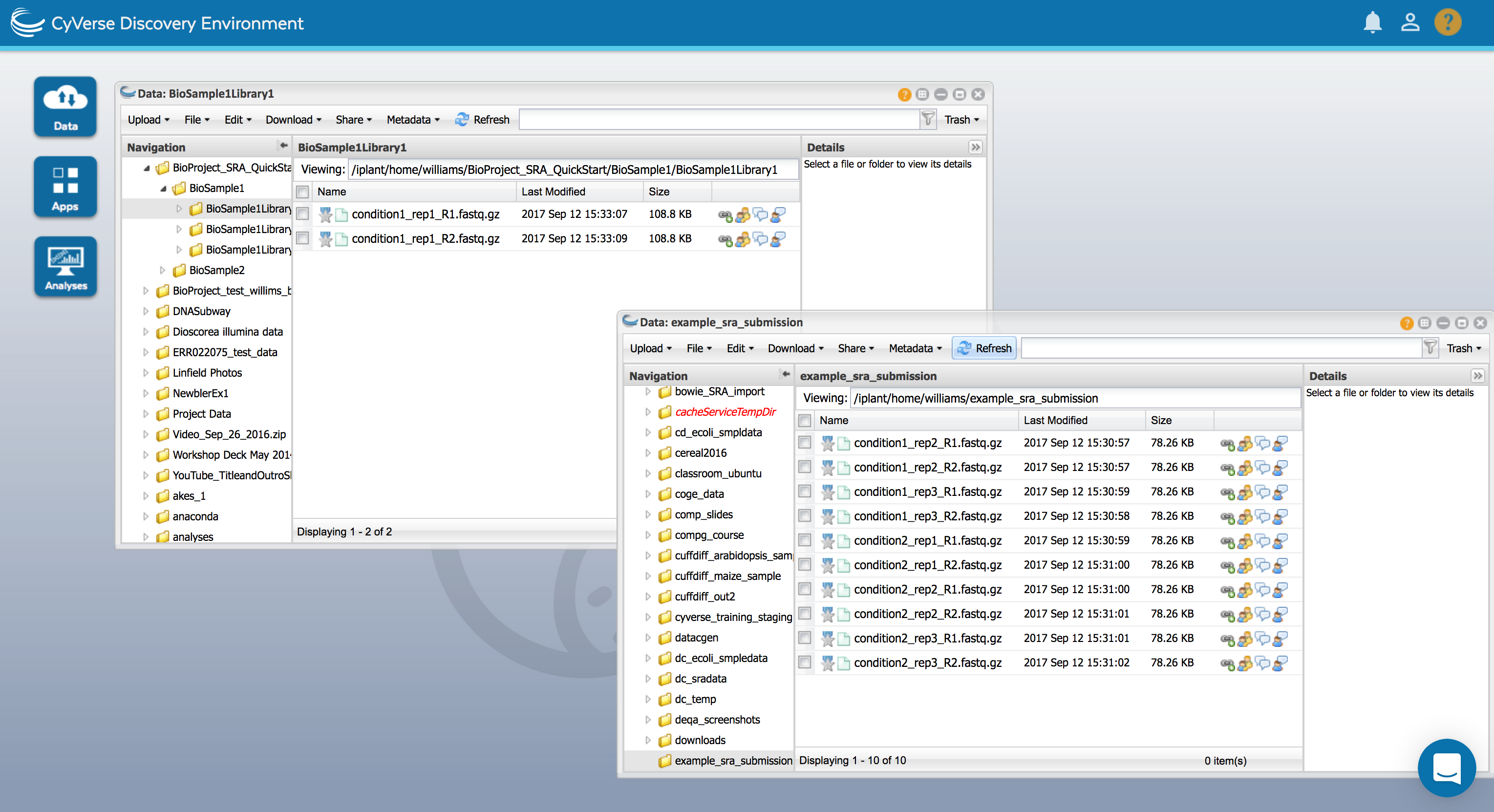
Place your sequence files in the appropriate library folders.
Tip
You can open multiple data window to facilitate drag-and-drop movement of files; from an open “Data” window click ‘file’ and select ‘New Data Window.’
Examine the example submission (BioProject_SRA_QuickStart folder) for refference and ensure your sequencing samples are appropriately organized.
Fix or improve this documentation
- On Github: Repo link
- Send feedback: Tutorials@CyVerse.org